L’équipe "chimie théorique et modélisation" (CTM) est un groupe dynamique de recherche en chimie computationnelle dont les activités s’organisent autour de thématiques variées allant de la molécule aux systèmes complexes en tenant compte de l’influence de l’environnement sur ses propriétés physiques ou chimiques.
Son domaine d’expertise, large, s’articule autour du développement et application de nouveaux protocoles de modélisations basés des méthodes de mécanique moléculaire et chimie théorique telles que la théorie de la fonctionnelle de la densité (DFT) et sa variante dépendant du temps (TDDFT). Ces approches, originales, sont appliquées sur une large palette d’objets d’études allant de la molécule aux surfaces en passant par les biomolécules, au plus près des problématiques expérimentales. A ce jour, les activités de recherches sont organisées autour de trois grandes thématiques.
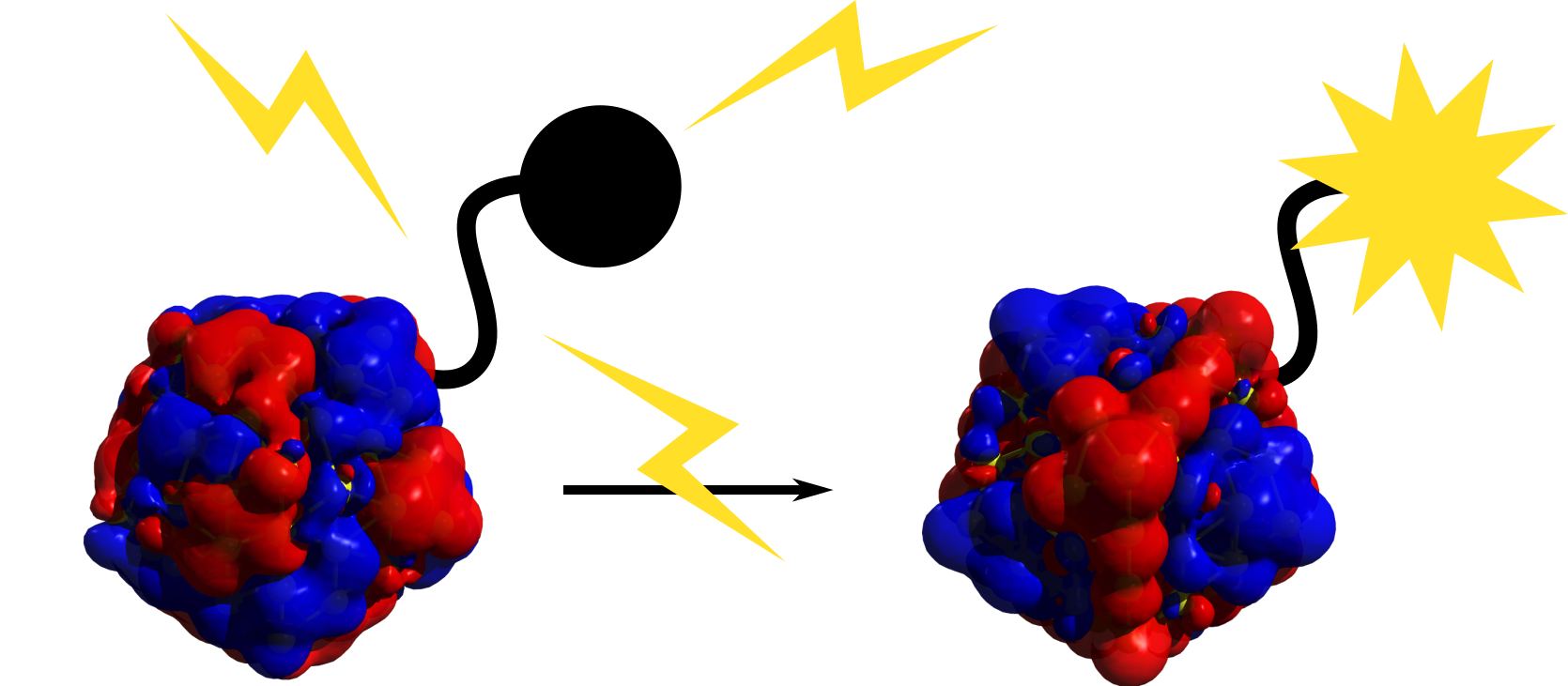 L’équipe CTM développe de nouvelles méthodologies en DFT pour l’étude des propriétés de l’état fondamental et de l’état excité de systèmes complexes dérivés de la plasmonique moléculaire ou à caractère photoactif, comme des photochromes conjugués greffés à la surface d’une nano-particule d’or. Ces développements permettent de comprendre la nature et l’importance des processus photodynamiques impliqués, ainsi que d’aller vers le design de systèmes photo-actifs pour les nanosciences.
L’équipe CTM développe de nouvelles méthodologies en DFT pour l’étude des propriétés de l’état fondamental et de l’état excité de systèmes complexes dérivés de la plasmonique moléculaire ou à caractère photoactif, comme des photochromes conjugués greffés à la surface d’une nano-particule d’or. Ces développements permettent de comprendre la nature et l’importance des processus photodynamiques impliqués, ainsi que d’aller vers le design de systèmes photo-actifs pour les nanosciences.
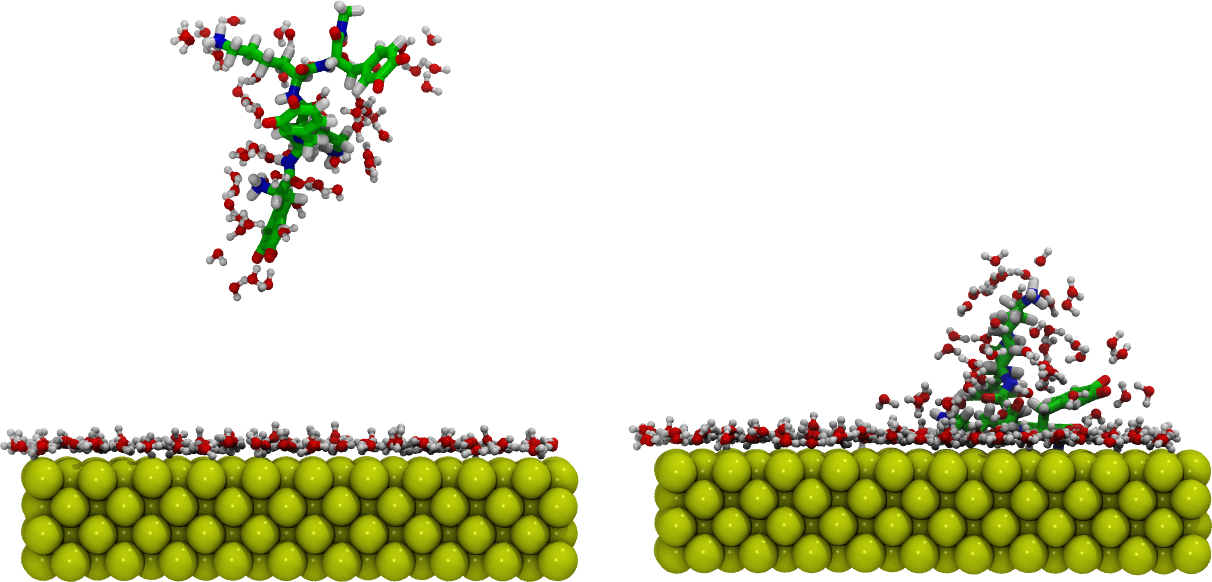 L’élucidation des interactions ligand/biomolécule est un axe de développement historique de l’équipe qui met en œuvre un panel important de techniques de simulations. Plus récemment, l’équipe CTM s’intéresse à l’étude des biomatériaux en appliquant des modélisations multi-échelles allant de la quantique à la mécanique et dynamique moléculaire. L’étude de l’assemblage supramoléculaire sur des surfaces est également menée dans l’équipe en utilisant notamment la DFT en condition périodique pour élucider le processus de nanostructuration ainsi que la modulation des propriétés électroniques et les mécanismes de transport électronique.
L’élucidation des interactions ligand/biomolécule est un axe de développement historique de l’équipe qui met en œuvre un panel important de techniques de simulations. Plus récemment, l’équipe CTM s’intéresse à l’étude des biomatériaux en appliquant des modélisations multi-échelles allant de la quantique à la mécanique et dynamique moléculaire. L’étude de l’assemblage supramoléculaire sur des surfaces est également menée dans l’équipe en utilisant notamment la DFT en condition périodique pour élucider le processus de nanostructuration ainsi que la modulation des propriétés électroniques et les mécanismes de transport électronique.
L’équipe CTM s’intéresse à l’étude de la fragilisation de matériaux, en développant des méthodologies originales avec une approche multi-échelle utilisant les calculs DFT à l’échelle atomique pour paramétrer des simulations aux échelles mésoscopiques (simulations Monte Carlo, dynamique moléculaire classique, dynamique de dislocation) et macroscopique (calculs de structure par éléments finis) dans le cadre de collaborations nationales et internationales. Dans ce thème, l’équipe s’intéresse également au stockage de l’hydrogène dans les nanomatériaux poreux pour les problématiques de l’énergie.